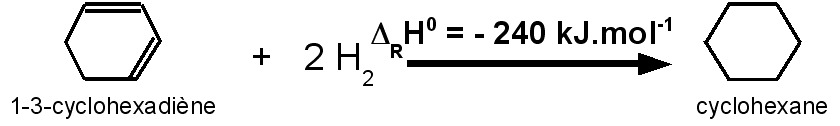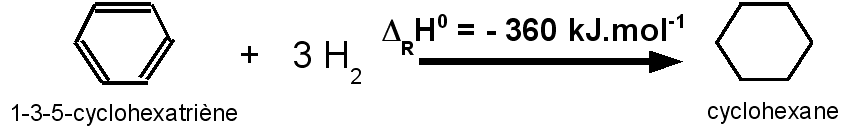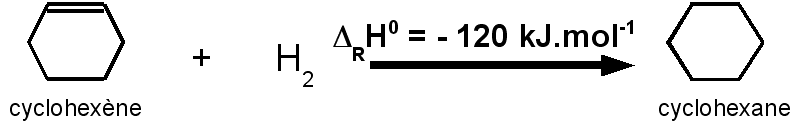
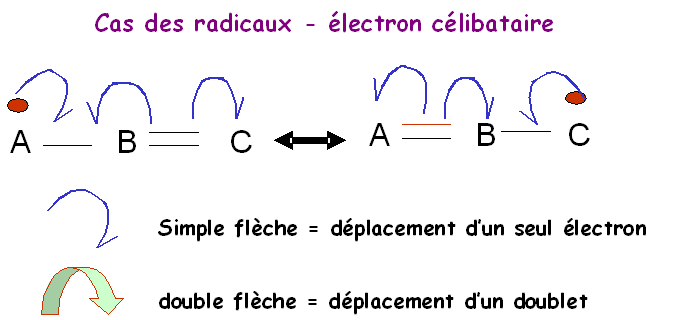
CHIM 321 – Bases de chimie organique – Cours de Thierry Brière – Mésomérie
Le phénomène de mésomérie est très
important en chimie organique car il modifie sensiblement les propriétés
chimiques des composés concernés.
Nous allons rappeler rapidement en quoi il
consiste.
Ce phénomène à déjà été traité en détail au premier
semestre.
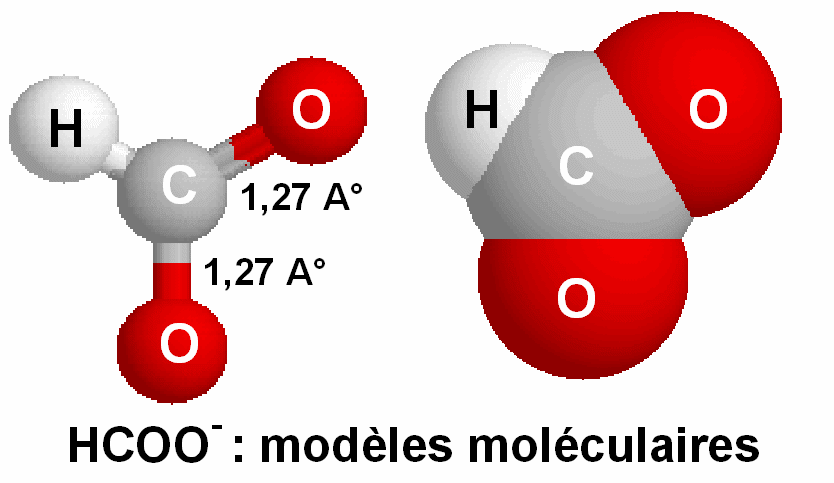
Considérons l'ion méthanoate
HCOO-
On peut facilement construire son schéma de Lewis :
Dans ce schéma on voit qu'une liaison CO est une
liaison double et l'autre liaison CO est une liaison simple.
On prévoit donc
à priori deux liaisons CO de longueurs différentes.
On peut d'ailleurs
estimer leurs valeurs par les formules empiriques :
R = 0,215
n2/Z* + 0,148 n + 0,225
dAB = 1,11 (RA +
RB ) - 0,203
Pour C : Z* = 3,25 et RC = 0,786
A°
Pour O : Z* = 4,55 et RO = 0,710 A°
dCO =
1,11*(0,786 + 0,710) - 0,203 = 1,458 A°
Soit finalement :
CO simple :
1,46 A°
CO double : 0,86 * 1,458 = 1,25 A°
La géométrie de l'ion méthanoate a put être déterminée expérimentalement, on constate que les deux longueurs de liaison CO sont rigoureusement identiques avec une valeur expérimentale de 1,27 A°.
|
|
|
Cette valeur est très proche de la
longueur prévue pour une double liaison CO.
Il semblerait donc que les deux
liaisons CO soient des liaisons doubles.
Cela n'est toutefois pas possible
puisque le carbone, élément de la deuxième période, ne possédant que 4 cases
quantiques sur sa couche de valence ne peut faire au maximum que 4 liaisons.
Comme il est également lié à l'hydrogène, il ne peut donc faire deux doubles
liaisons avec les atomes d'oxygène.
Pour expliquer simplement ce
phénomène on utilise le modèle de la mésomérie.
Il suffit de considérer
la symétrie de la molécule, les deux atomes d'oxygène n'ont aucune raison d'être
différents l'un de l'autre. Or, dans le schéma de Lewis précédant non seulement
ils ne feraient pas le même nombre de liaisons avec le carbone mais de plus l'un
serait porteur d'une charge négative et de trois doublets électroniques libres
et l'autre serait non chargé et ne posséderait que deux doublets électroniques
libres.
Mais il est facile de construire un deuxième schéma de Lewis
pour lequel les deux oxygènes échangent simplement leurs rôles. On passe
simplement d'une forme à l'autre par déplacement de doublets
électroniques.
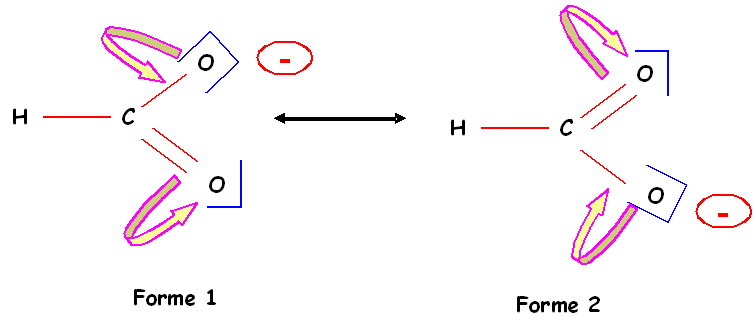
Ces deux formes sont strictement équivalentes l'une à l'autre, on dit qu'elles ont un poids statistique identique (revoir cours du premier semestre).
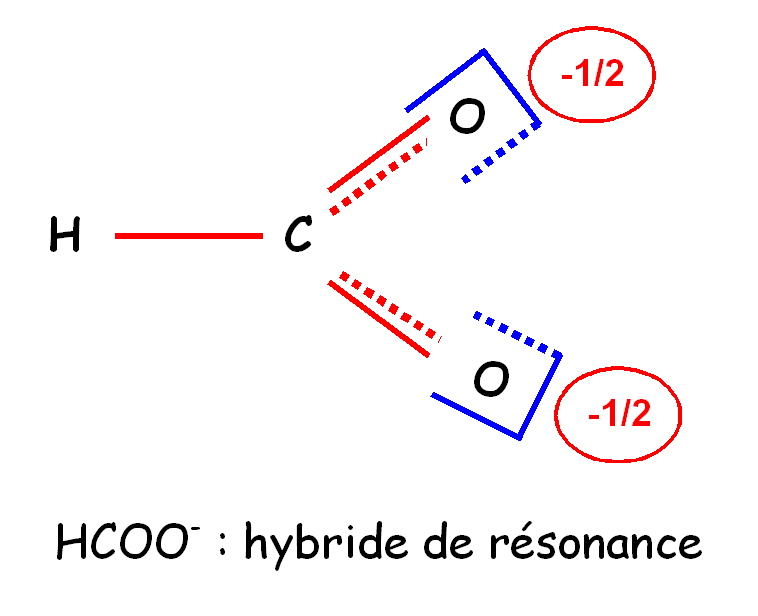
La molécule réelle sera un hybride de
résonance entre ces deux formes mésomères, dans cet hybride, les deux liaisons
et les deux oxygènes seront strictement équivalents entre eux.
Les doublets
électroniques qui se déplacent pour passer d'une forme à l'autre correspondent
au fait que les électrons dans un tel système ne sont pas strictement localisés.
Ils sont susceptibles de se déplacer dans la molécule, on dit qu'ils sont
délocalisables. C'est cela le phénomène de mésomérie. On les représentera par
des traits pointillés dans l'hybride de résonance. De même la charge électrique
négative n'est pas localisée sur un des atomes d'oxygène en particulier, elle se
répartit également entre les deux, on indiquera donc une charge -1/2 pour chaque
oxygène dans l'hybride de résonance.
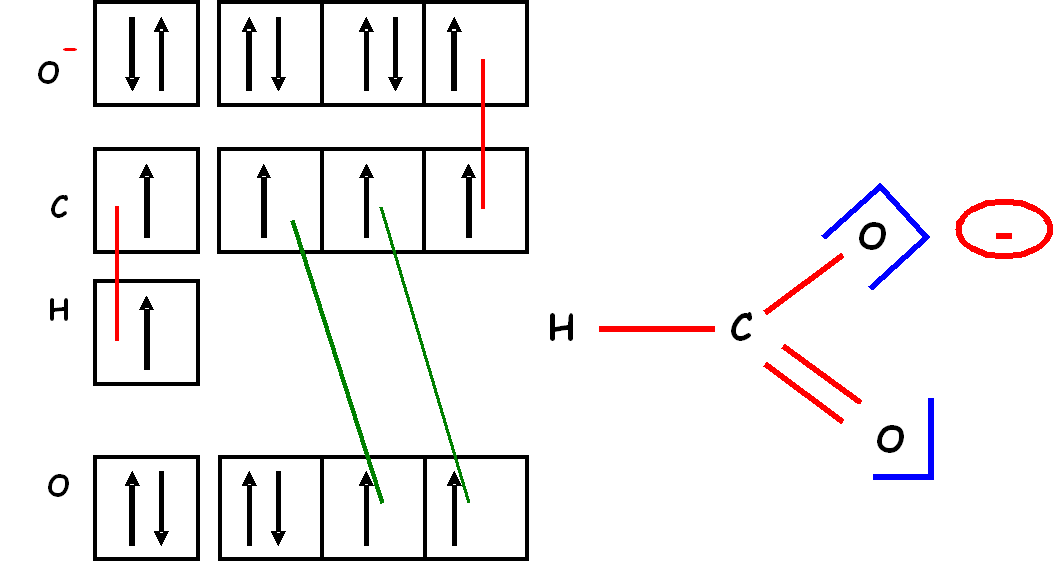
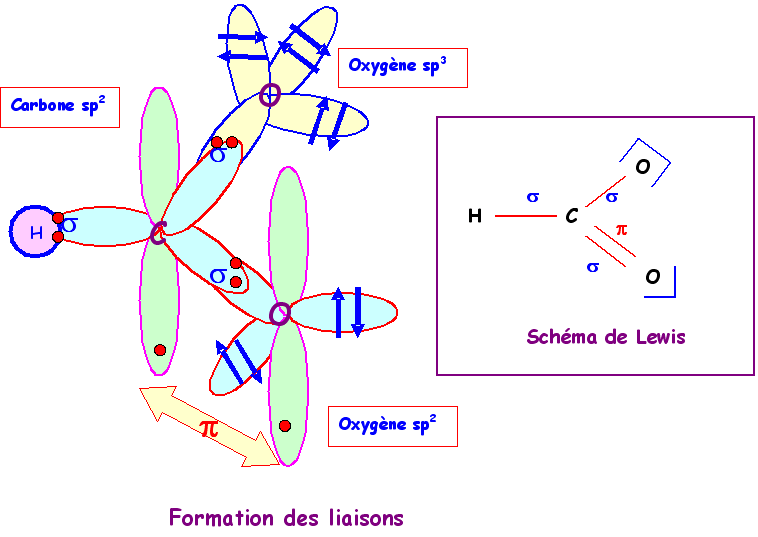
Si on s'en tient toujours au schéma de Lewis simple établi précédemment on peut prévoir les états d'hybridation des atomes permettant de le décrire.
L'atome de carbone est du type AX3 soit sp2.
L'atome d'oxygène porteur des trois doublets et de la charge négative est du type AXE3 soit une hybridation sp3.
L'autre oxygène porteur de deux doublets est du type AXE2 soit sp2.
Si on s'en tient à ces hypothèse on peut construire la molécule et justifier la formation des liaisons et . Soit le schéma suivant :
Cette description n'est toutefois pas
satisfaisante pour les même raisons de dissymétrie, les deux oxygènes sont
différents ce qui n'est pas acceptable.
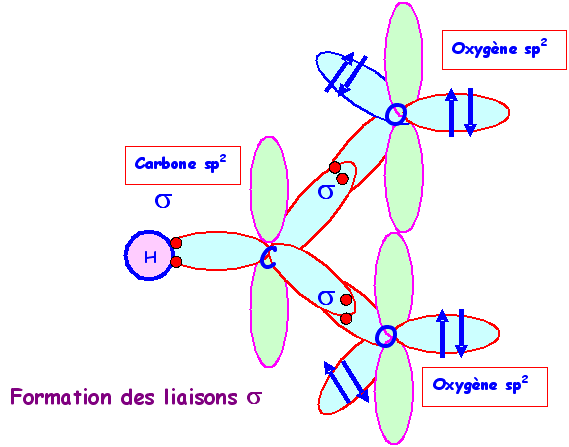
Pour obtenir une description
correcte nous allons utiliser deux atomes d'oxygène dans le même état
d'hybridation. Puisque la molécule réelle semble posséder deux doubles liaisons
nous utiliserons des oxygènes sp2 qui permettent d'obtenir les
liaisons
nécessaires.
Nous allons voir que cette description justifiera simplement
l'existence des deux formes mésomères précédemment décrites.
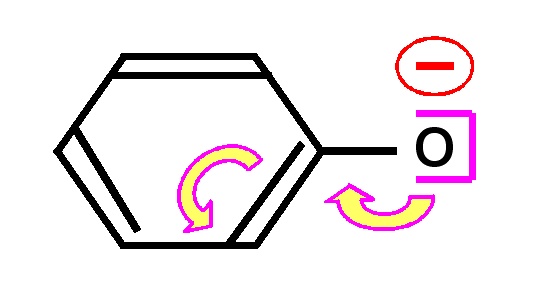
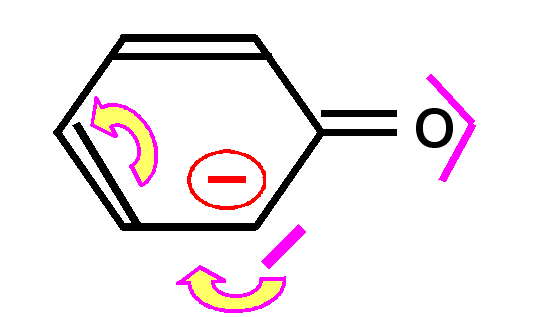
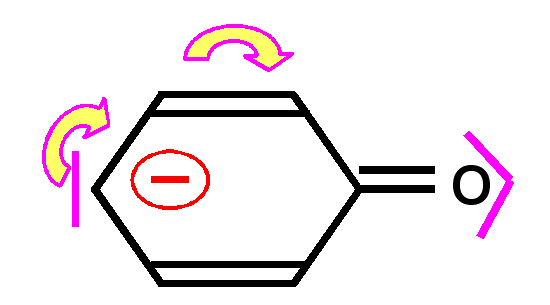
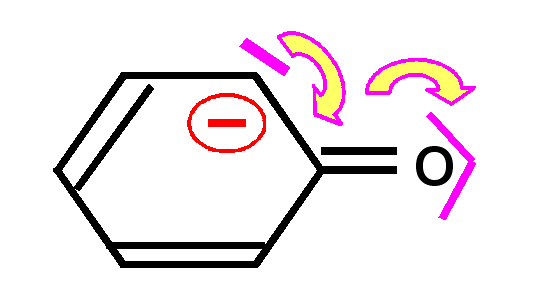
En effet, la liaison pourra se faire de deux manières différentes conduisant aux deux formes mésomères.
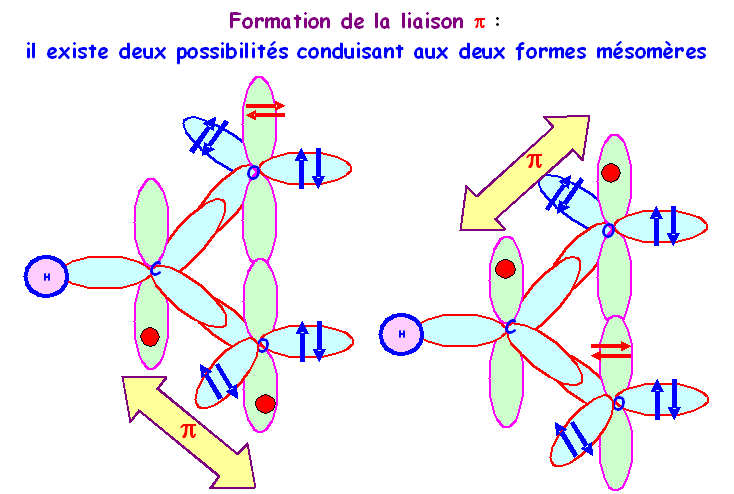
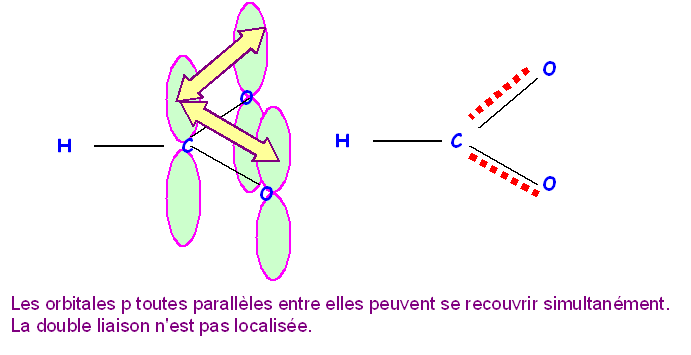
Ce phénomène de délocalisation des électrons des orbitales p formant des liaisons est tout à fait général et on le retrouvera à chaque fois que deux ou plusieurs atomes en état d'hybridation sp2 seront liés entre eux.
Dans les composés organiques le cas le plus courant est l'alternance de doubles et de simples liaisons. On dit alors que les doubles liaisons sont conjuguées entre elles.
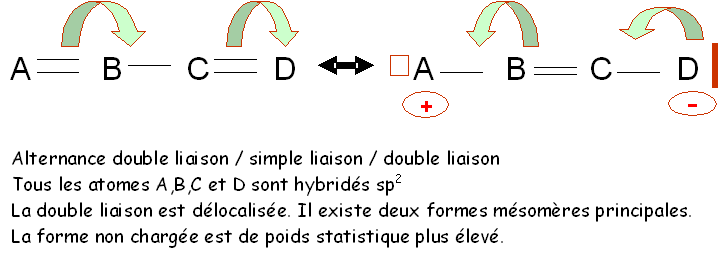
Remarque :
Si un atome hybridé sp3 vient s'intercaler, la conjugaison n'existe plus et les électrons ne sont plus délocalisés
CH2=CH-CH2-CH=CH2 : sp2 - sp2 - sp3 - sp2 - sp2 : doubles liaisons non conjuguées, électrons non délocalisés.
Le schéma de Lewis de la molécule montre que la
géométrie est du type AX3 autour du carbone et
AX2E2 autour de l'oxygène porteur de l'atome d'hydrogène.
Enfin, on peut considérer qu'elle est du type AXE2 autour de
l'autre atome d'oxygène.
On a donc des hybridations du type
:
sp2 pour C
sp3 pour le O de OH
sp2 pour le O de C=O
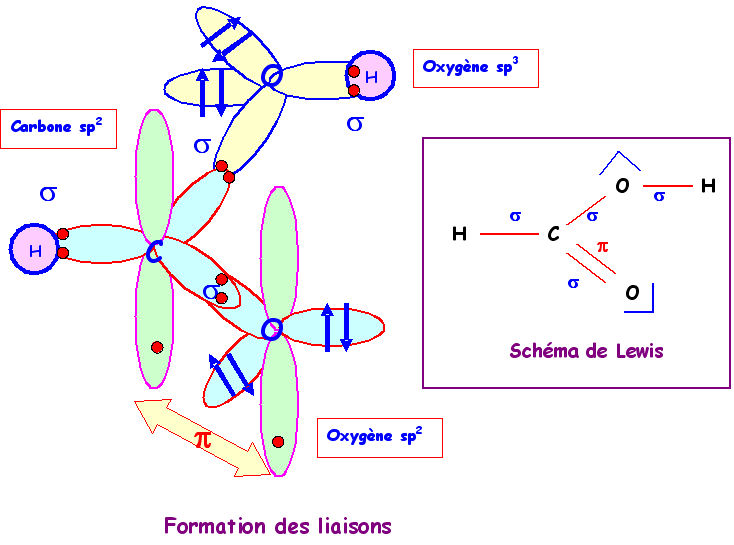
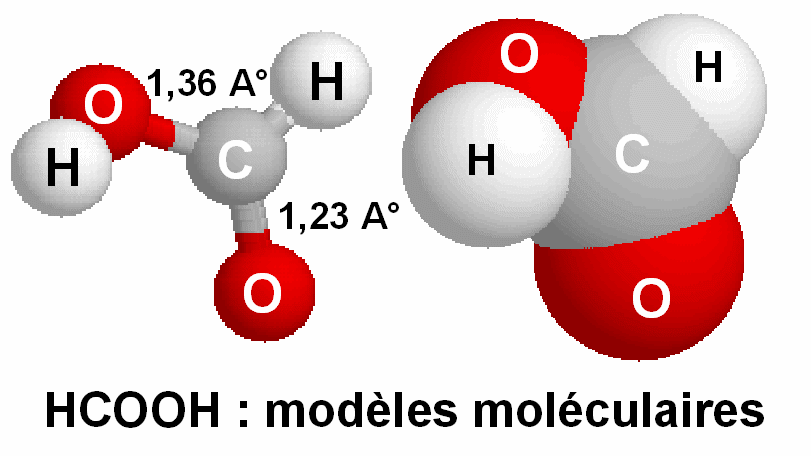
Cette molécule a été étudiée
expérimentalement, les longueurs de liaison CO observées sont respectivement
1,23 A° pour C=O et 1,36 A° pour C-OH.
|
|
|
Comparons ces valeurs avec les valeurs
estimées calculées précédemment :
C-O : 1,46 A° et C=O : 1,25
A°
Pour la double liaison, la valeur estimée et la valeur expérimentale
sont en bon accord avec1,6 % d'écart seulement. 100 * (1,25 - 1,23) / 1,23 =
1,6
Pour la simple liaison, l'écart observé est de 7,5 % et l'accord est donc
très mauvais.
100 * ( 1,46 - 1,36 ) / 1,36 = 7,4
Il semble que la liaison
C-O est anormalement courte. Comment expliquer cela ?
Ici aussi c'est l'effet
mésomère qui intervient et raccourcit cette liaison.
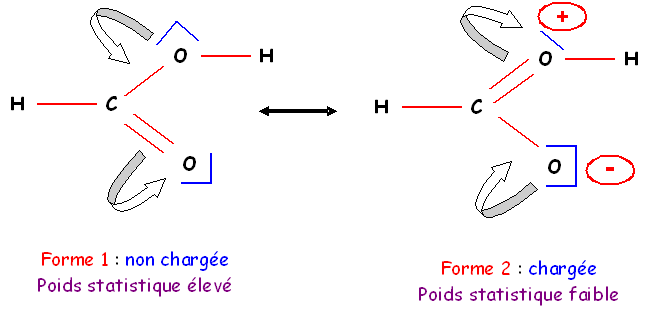
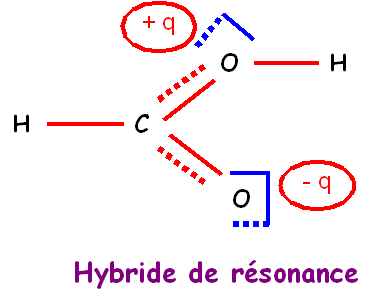
La forme 2 chargée a un poids statistique
faible, mais dans l'hybride de résonance la liaison C-O prend un caractère de
liaison partiellement double et sa longueur est donc plus courte que prévu pour
une liaison simple.
Les deux formes ayant des poids statistiques très
différents l'hybride de résonance reste très proche de la forme 1. Les charges
partielles q portées par les deux oxygènes sont très faibles. Dans la pratique
seule la longueur de la liaison simple est sensiblement raccourcie. Cela est
très général, on observe très souvent ce cas de figure.
L'existence de la
forme 2 justifie donc les longueurs de liaisons expérimentales, il présente en
outre l'intérêt d'expliquer l'acidité des acides carboxyliques.
En effet, la
densité électronique est fortement abaissée sur l'atome d'oxygène positif.
L'oxygène très électronégatif va chercher à récupérer des électrons pour
compenser. Il va donc se tourner vers l'hydrogène voisin pour lui prendre son
électron.
La liaison
sera alors rompue et on obtiendra deux ions H+ et
HCOO-.
Cette ionisation est de plus favorisée par le fait que
l'ion carboxylate HCOO- obtenu est lui même fortement stabilisé par
mésomèrie comme nous l'avons vu plus haut.
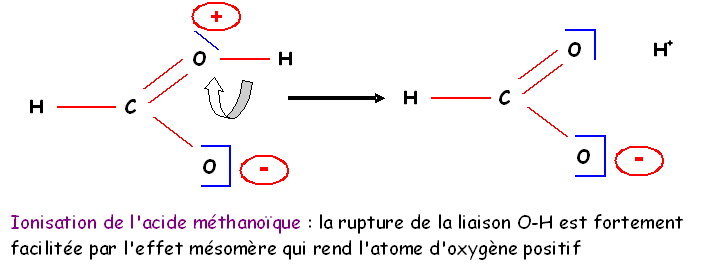
Dans cet exemple l'effet mésomère concerne un doublet libre de l'oxygène conjugué à une double liaison. On rencontre fréquemment ce cas de figure.
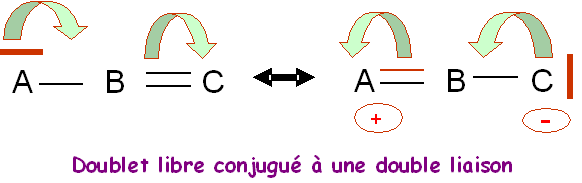
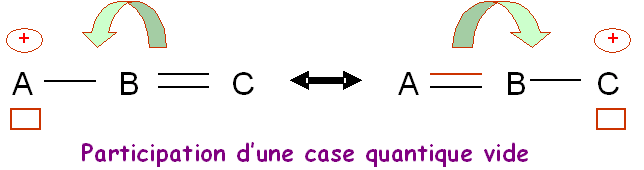
Le benzène est un cas
typique de molécule conjuguée.
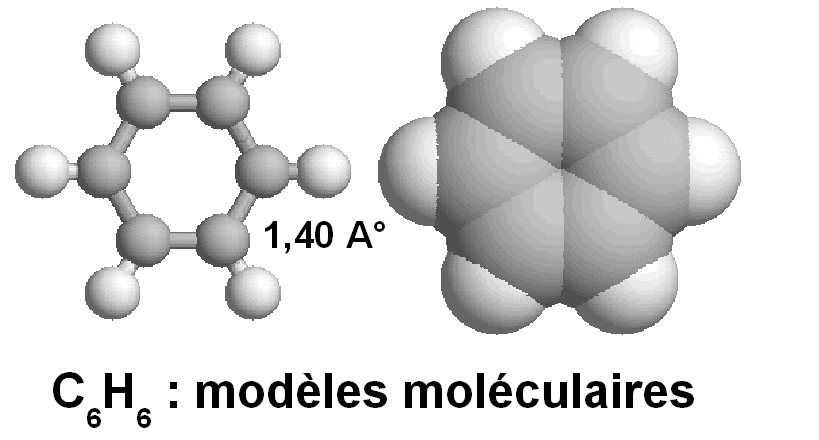
Sa géométrie moléculaire établie
expérimentalement montre que les 6 atomes de carbone sont situés aux sommets
d'un hexagone régulier.
Les six atomes d'hydrogène sont dans le même plan que
les carbones.
Les liaisons CC sont identiques et de longueur 1,40
A°.
Comparons cette longueur avec les longueurs prévues pour des liaison CC
simples et CC doubles :
RC = 0,786 A°
dCC simple =
1.11 * 2 * 0,786 - 0,203 = 1,54 A°
dCC double = 0,86 * 1,54
= 1,33 A°
La longueur expérimentale 1,40 A° est donc
intermédiaire entre simple et double liaison.
|
|
|
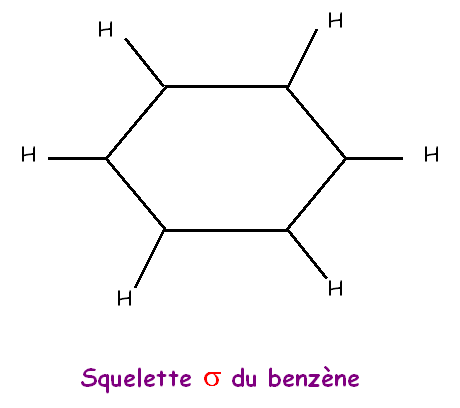
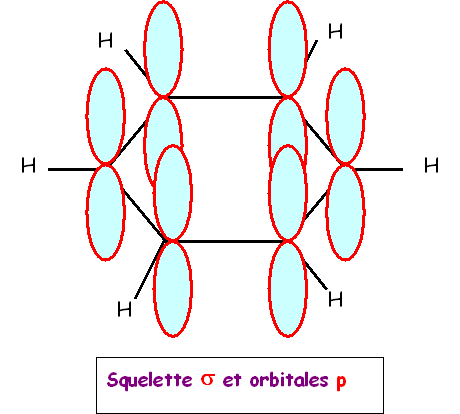
La géométrie moléculaire observée s'accorde parfaitement avec une hybridation sp2 des 6 atomes de carbone de type AX3.
On peut facilement en utilisant ce type
d'hybridation construire le squelette sigma du benzène dans le modèle
quantique.
Les orbitales sp2 pointant à 120° les unes des
autres conduisent naturellement à la géométrie hexagonale observée.
Tous
les atomes C et H seront bien situés dans un même plan.
Il restera des orbitales p non utilisées qui pourront former les doubles liaisons pi.
|
|
|
Les orbitales p étant toutes parallèles
entre elles, on aura plusieurs possibilités de recouvrement différentes, ce qui
conduira aux diverses formes mésomères.
On décrit les orbitales p comme
formant un nuage électronique englobant tous les atomes de carbone, on dit qu'il
y a délocalisation des électrons p.
|
|
|
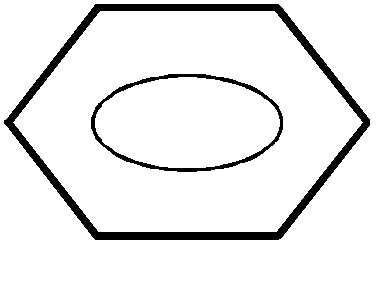
Finalement on adopte une représentation
symbolique de la molécule de benzène tenant compte de cette délocalisation des
électrons.
On ne représentera pas les liaisons doubles comme localisées.
Elles seront figurées par un cercle à l'intérieur de
l'hexagone.
La conjugaison
entraîne une stabilisation importante des molécules.
Plus une molécule
possède de formes mésomères et plus celle-ci est stable.
On définit cette
stabilisation par l'énergie de résonance, nous verrons en T.D des exemples de
détermination de ces énergies de résonance par voix thermodynamiques.
On peut
aussi les déterminer théoriquement grâce à la mécanique quantique, mais cela
dépasse le niveau élémentaire de ce cours…
Prenons l'exemple du benzène pour
fixer les idées.
Une façon simple d'évaluer la stabilité des molécules de ce
type est de comparer leurs enthalpies d'hydrogénation.
L'hydrogénation du cyclohexène C6H10 pour conduire au cyclohexane C6H12 a une enthalpie de DRH0 = -120 kJ.mol-1.
L'hydrogénation du cyclohexa-1,3-diène C6H8 pour conduire au cyclohexane C6H12 a une enthalpie de DRH0 = -230 kJ.mol-1.
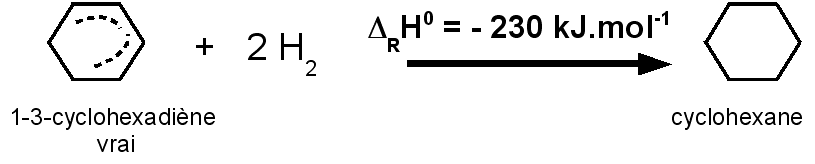
L'hydrogénation du benzène C6H6 pour conduire au cyclohexane C6H12 a une enthalpie de DRH0 = -210 kJ.mol-1.
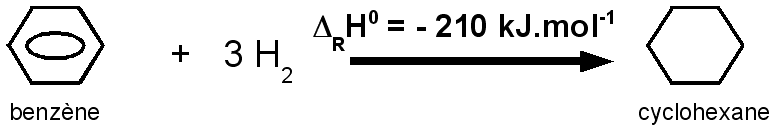
Comparons ces valeurs entre elles.
L'hydrogénation du cyclohexène correspond à celle d'une seule double liaison C=C.
L'hydrogénation du cyclohexadiène correspond à celle de deux double liaison C=C.
L'hydrogénation du benzène correspond à celle de trois double liaison C=C.
On peut supposer que grossièrement l'hydrogénation d'une double liaison C=C doit avoir une enthalpie sensiblement constante.
Si on se base sur la valeur de 120 kJ.mol-1 pour une double liaison C=C :
l'hydrogénation du
cyclohéxadiène devrait correspondre à environ :
2 * 120 = 240
kJ.mol-1.
l'hydrogénation du
benzène devrait correspondre à environ :
3 * 120 = 360
kJ.mol-1.
L'accord semble assez
correct pour le cyclohexadiène avec un écart de seulement 10
kJ.mol-1.
En revanche pour le benzène il n'en est rien et on
observe un écart de 150 kJ.mol-1.
Pour faire cette
évaluation simple, on suppose que les doubles liaisons sont purement
individuelles et parfaitement localisées.
Mais nous savons qu'il n'en est
rien et qu'elles sont en réalité conjuguées entre elles.
Par conséquent, il
est incorrect de dire que le cyclohexadiène réel correspond simplement à deux
doubles liaisons isolées.
La valeur calculée de 2 * 120 = 240
kJ.mol-1 correspond à celle d’un cyclohexadiene hypothétique à deux
doubles liaisons localisées.
|
|
|
|
L'écart observé de 10 kJ.mol-1 correspond à l'énergie de résonance des deux doubles liaisons conjuguées.
Energie de
résonance du cyclohexadiene: ER = 240 –
230 = 10 kJ.mol-1
De même, le benzène réel ne
correspond pas à trois doubles liaisons isolées et localisées.
La valeur
calculée de 3 * 120 = 360 kJ.mol-1 correspond à celle d’un
cyclohexatriene hypothétique à 3 doubles liaisons localisées.
|
|
|
|
L'écart observé de 150
kJ.mol-1 correspond à l'énergie de résonance des trois doubles
liaisons conjuguées.
Energie de résonance du benzène :
ER = 360 – 210 = 150
kJ.mol-1
Ces enthalpies d'hydrogénation permettent de
comparer les enthalpies standard de formation des divers
composés.
Exemple : Hydrogénation du cyclohexène
|
DfH0cyclohexène = DfH0cyclohexane - DRH01 |
Pour
alléger quelques peu les écritures nous prendrons une référence arbitraire en
posant que l'enthalpie standard de formation du cyclohexane est considérée comme
nulle. L'enthalpie standard de formation du dihydrogène H2 est elle
aussi nulle par définition.
Avec ces simplifications, l'enthalpie de la
réaction d'hydrogénation est une mesure directe de l'enthalpie standard de
formation du composé subissant l'hydrogénation.
On pourra donc comparer la
stabilité des divers composés.
|
DfH0cyclohexène = DfH0cyclohexane - DRH01 DfH0cyclohexène = - DRH01 = 120 kJ.mol-1 |
|
DfH0cyclohexadiène = DfH0cyclohexane - DRH03 DfH0cyclohexadiène = - DRH03 = 240 kJ.mol-1 |
|
DfH0cyclohexadiène vrai = - DRH04 = 230 kJ.mol-1 |
|
DfH0cyclohexatriène = DfH0cyclohexane – DRH05 DfH0cyclohexatriène = - DRH05 = 360 kJ.mol-1 |
|
DfH0benzène = DfH0cyclohexane - DRH05 DfH0benzène = - DRH05 = 210 kJ.mol-1 |
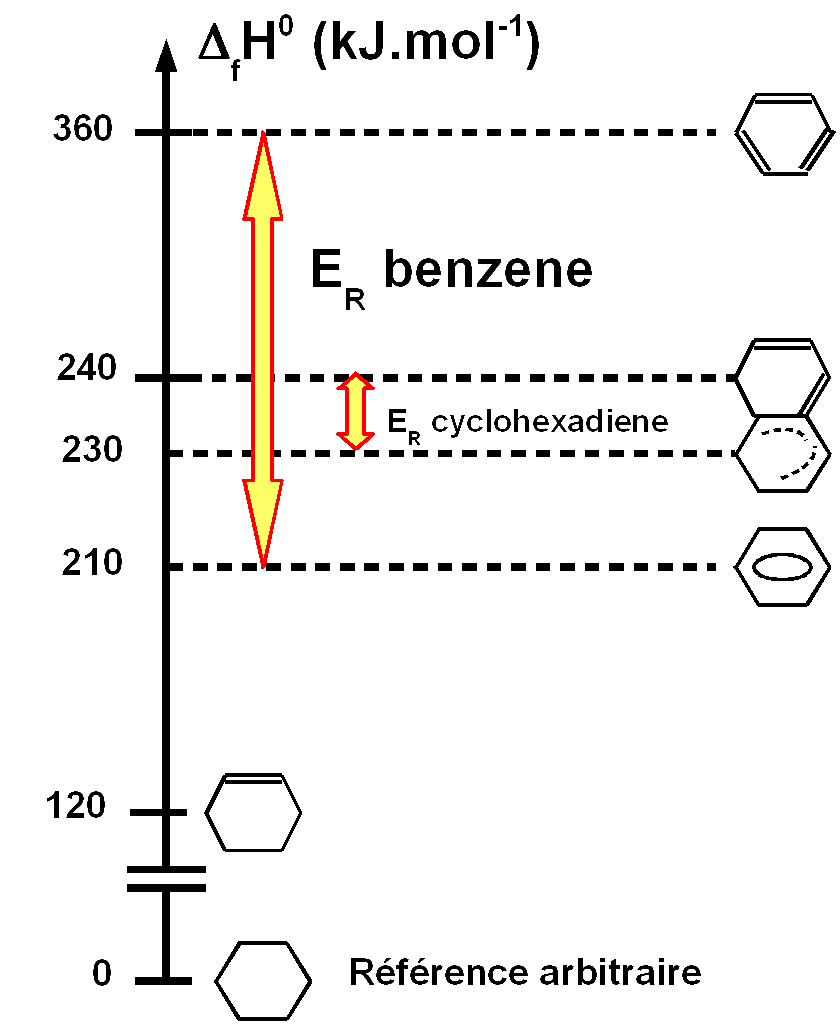
On voit
sur le diagramme la très grande stabilisation par résonance du
benzène.
Le phénomène de conjugaison stabilise très
fortement la molécule de benzène, son enthalpie de formation est beaucoup plus
basse que celle de la molécule hypothétique non conjuguée.
L'effet est
beaucoup moins important pour la molécule de cyclohexadiene.
Ces résultats
sont généraux, l'effet mésomère stabilise d'autant plus fortement les molécules
que le nombre de doubles liaisons conjuguées est important.
Remarques :
On peut vérifier expérimentalement que l'enthalpie d'hydrogénation d'une double liaison est bien sensiblement constante et vaut environ -122 kJ.mol-1.
On peut vérifier également que pour un diène non conjugué l'enthalpie d'hydrogénation est bien sensiblement le double de celle d'une seule double liaison.
|
Composé |
Enthalpie d'hydrogénation kJ.mol-1 |
Nombre de C=C |
DH moyen pour 1 C=C kJ.mol-1 |
|
CH3-CH2-CH=CH2 |
- 127 |
1 |
- 127 |
|
E - CH3-CH2-CH=CH2 |
- 115 |
1 |
- 115 |
|
Z - CH3-CH2-CH=CH2 |
- 119 |
1 |
- 119 |
|
CH2=CH-CH2-CH2-CH=CH2 |
- 253 |
2 |
- 126 |
|
CH2=CH-CH2-CH=CH2 |
- 254 |
2 |
- 127 |
|
cyclohexène |
- 120 |
1 |
- 120 |
|
|
|
|
Valeur moyenne : - 122 |
On peut facilement évaluer les énergies de résonance par calcul des enthalpies de formation des formes hypothétiques à liaisons localisées à partir des énergies de liaison et comparaison avec la molécule réelle. Nous traiterons des exemples en travaux dirigés.
On classe les groupements d’atomes (groupe fonctionnels) possédant un effet mésomère en deux catégories :
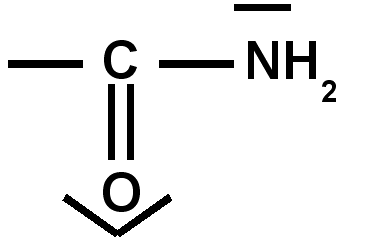
Groupes à effet mésomère donneur + M : ce sont généralement des groupes « riches en électrons » comportant un atome possesseur d’un doublet libre.
|
| |||||
|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
chloro |
fluoro |
alcool |
ether |
amine |
alcoolate |
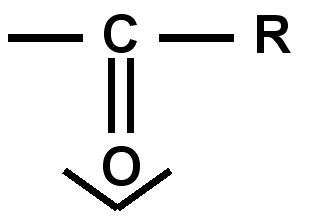
Groupes à effet mésomère accepteur - M : ce sont généralement des groupes « pauvres en électrons » comportant une ou plusieurs doubles liaisons ou atome sp2 ou cases quantiques vides.
|
| ||||||
|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
amide |
ester |
cétone |
aldéhyde |
nitrile |
sulfonique |
nitro |
Ces divers groupements seront étudiés en détail ultérieurement.
Nous avons expliqué plus haut l'acidité du groupement - COOH par mésomérie.
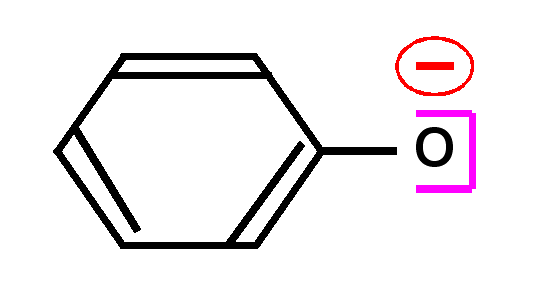
Acidité des phénols : La fonction phénol pour laquelle
les doublets de l'atome d'oxygène sont en conjugaison avec le noyau benzénique
sont beaucoup plus acides que les alcools ordinaires. Cela s'explique entre
autre par la stabilisation par mésomérie de l'ion phénate.
Le pKa des
phénols est de l'ordre de pKa =10, alors que celui des alcools ordinaires est
de l'ordre de pKa = 20.
|
|
|
|
|
|
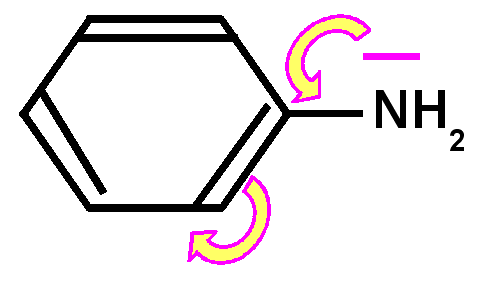
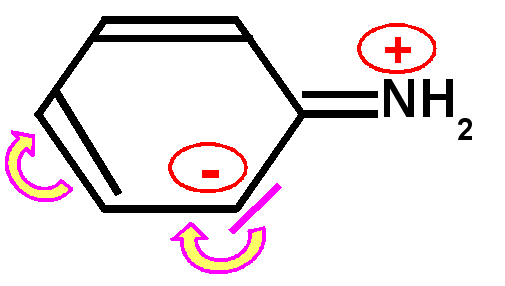
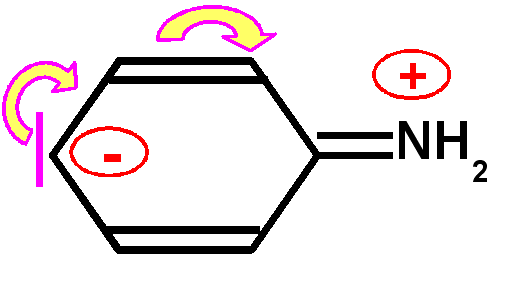
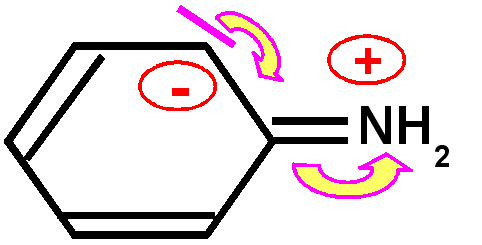
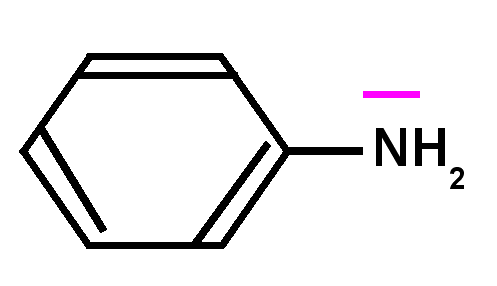
Basicité des amines : Une amine conjuguée sera moins basique qu'une amine non conjuguée, le doublet libre de l'azote responsable de la basicité selon Lewis sera en effet mis en jeu par la conjugaison et sera donc moins disponible d'ou une basicité plus faible et un pKa nettement plus petit.
Exemple de l'aniline : Le pKa de l'aniline est pKa = 4,6 à comparer avec l'ammoniaque NH3(aq) dont le pKa est de 9,2.
|
|
|
|
|
|
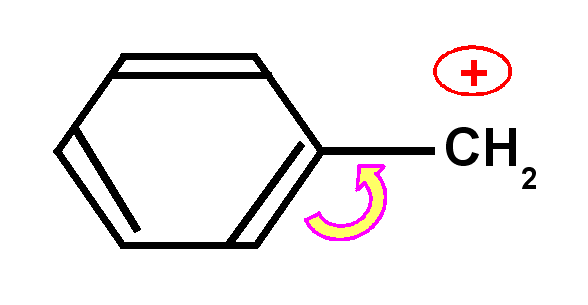
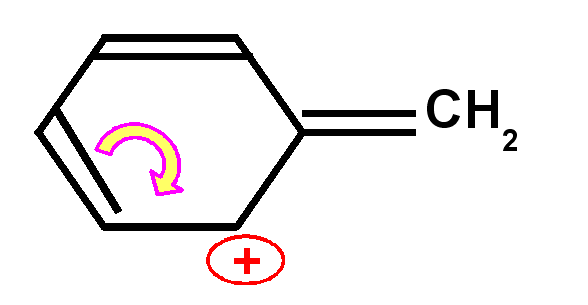
Les
carbocations et les carbanions peuvent être fortement stabilisés par
mésomérie.
La mésomérie permet de répartir la charge sur plusieurs atomes ce
qui stabilise grandement la structure.
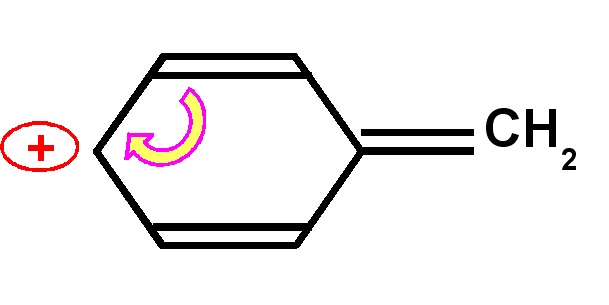
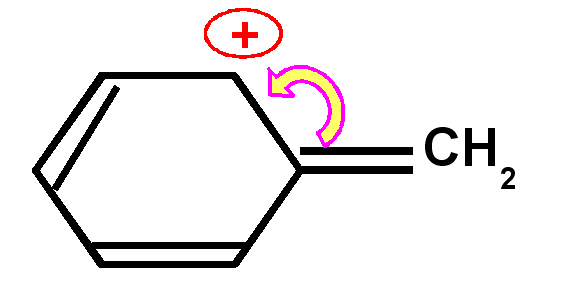
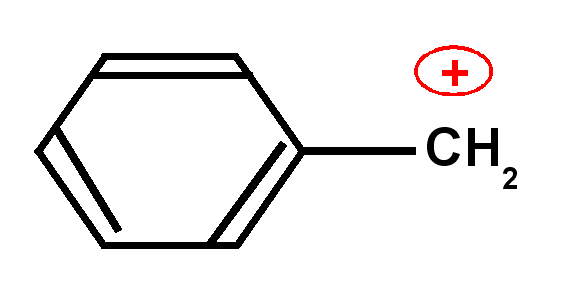
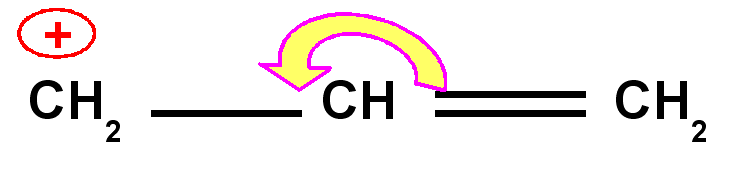
Carbocation benzylique
|
|
|
|
|
|
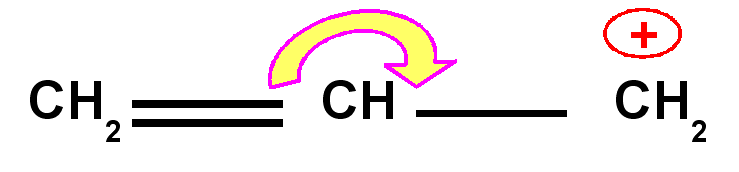
Carbocation allylique
|
|
|
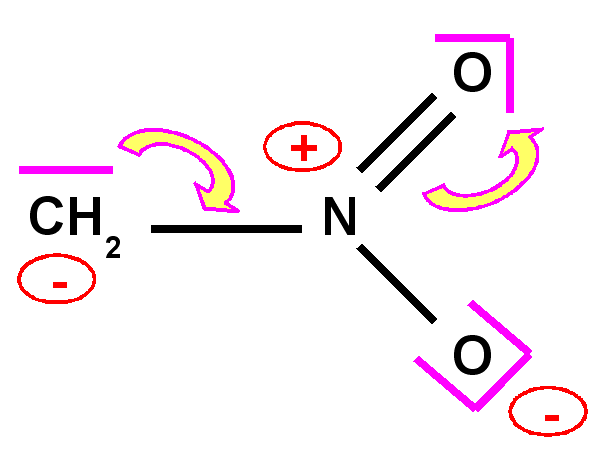
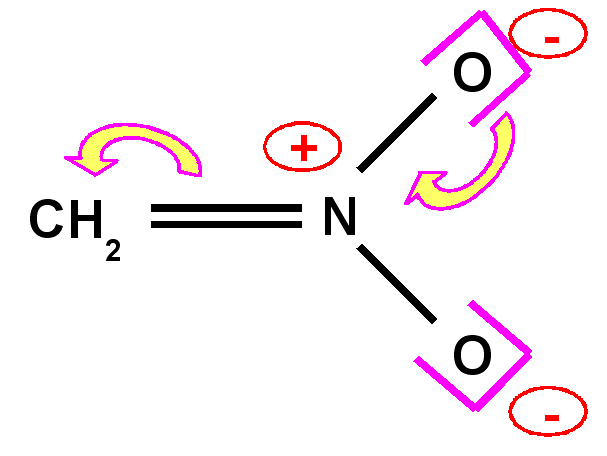
Carbanion nitrométhane
|
|
|
L'effet mésomère en déplaçant les charges électriques a l'intérieur d'une molécule peut orienter une réaction chimique sur un site particulier. Il peut rendre un site plus réactif, ou inversement inactiver celui-ci.
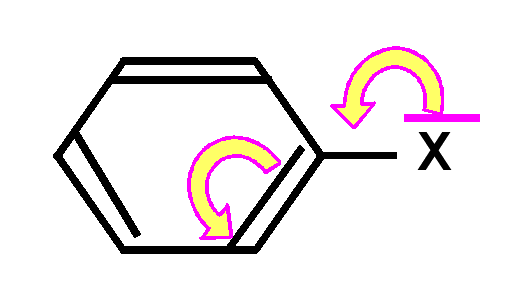
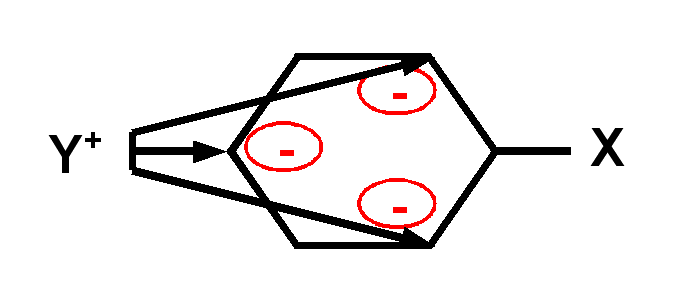
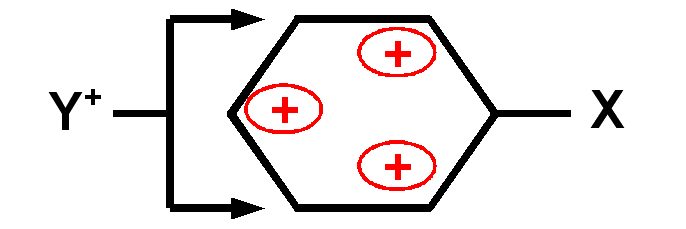
Exemple réaction de substitution nucléophyle aromatique :
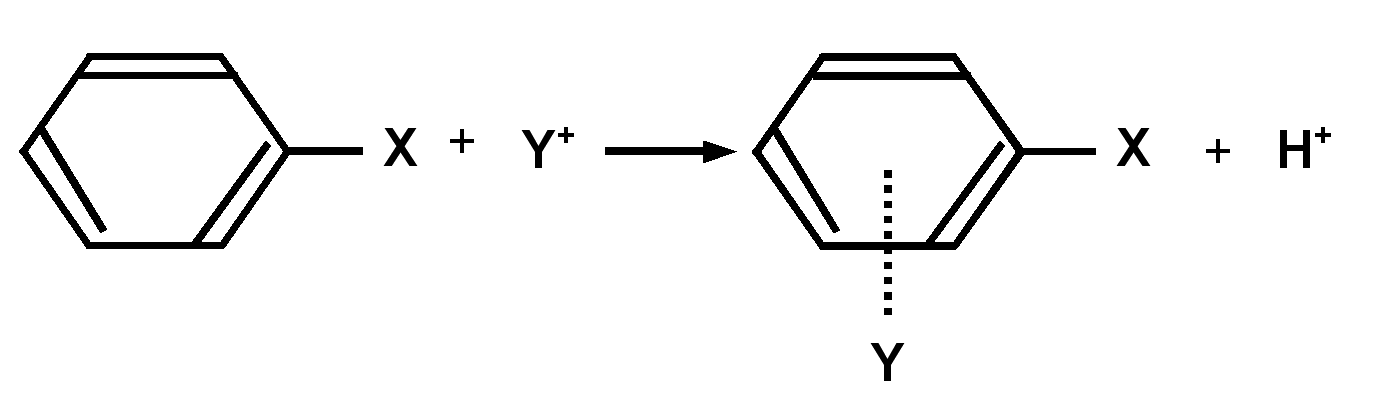
Dans ce type de réaction, un nucléophyle
Y+ vient se fixer sur le noyau benzénique en remplaçant un hydrogène
expulsé sous forme de proton H+.
On constate qu'avec un groupement
X électrodonneur (effet +M), la réaction est assez rapide et que la substitution
se fait préfferentiellement en positions ortho et para.
Inversement, avec un
groupement X électroattracteur (effet -M), la réaction est très lente et la
substitution se fait préfferentiellement en position méta.
Cela s'explique
par les effets mésomères des groupement X :
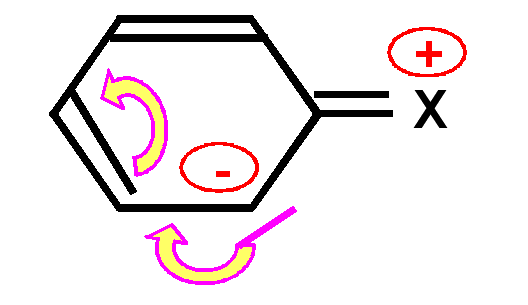
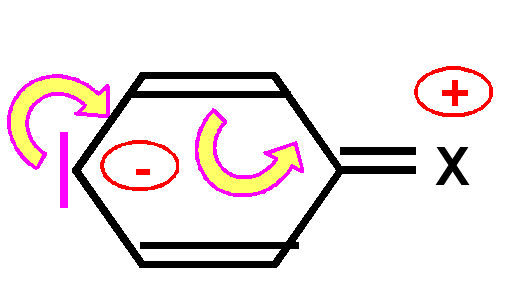
un groupe X électrodonneur enrichit le noyau aromatique en électrons en faisant apparaître des charges négatives sur les positions ortho et para. Cela oriente la substitution sur ses positions privilégiées et facilite celle-ci.
|
|
|
|
|
|
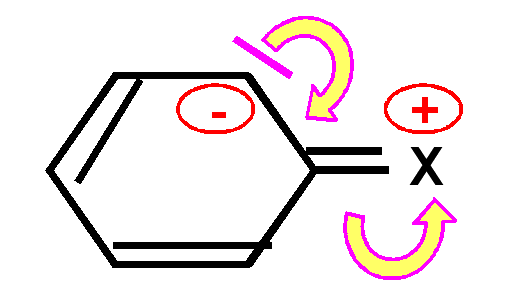
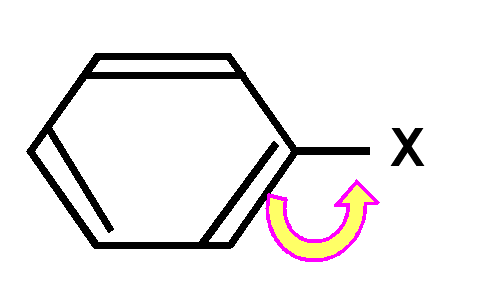
un groupe X électroattracteur apauvrit le noyau aromatique en électrons en faisant apparaître des charges positives sur les positions ortho et para. Cela rend la réaction très difficile et l'oriente plutôt sur les positions méta non apauvries en électrons.
|
|
|
|
|
|