LA GEOMETRIE DES MOLECULES
FORME GEOMETRIQUE DES MOLECULES - METHODE V.S.E.P.R (ou R.P.E.C.V)
Les molécules possèdent une certaine forme géométrique. Les liaisons autour de l'atome central ont une certaine orientation qui va donner une forme particulière à la molécule. L'objet de ce chapitre va être la prévision à priori de la forme d'une molécule. Le problème ne se pose que pour les molécules comportant au moins trois atomes, en effet les molécules diatomiques seront forcément linéaires puisque deux points ne peuvent qu'être alignés. A partir de trois atomes ont va pouvoir obtenir plusieurs géométries différentes selon les angles que feront les liaisons entre elles.
Principe de la Méthode V.S.E.P.R
Cette méthode mise au point par Gillespie permet de prévoir très simplement la forme géométrique des molécules à partir de leur schéma de Lewis moléculaire.
Le sigle V.S.E.P.R signifie en anglais " Valence Schell Electronic Pairs Répulsion ", ce qui traduit en français donne " Répulsion des Paires Electroniques de la Couche de Valence" soit R. P. E. C.V.
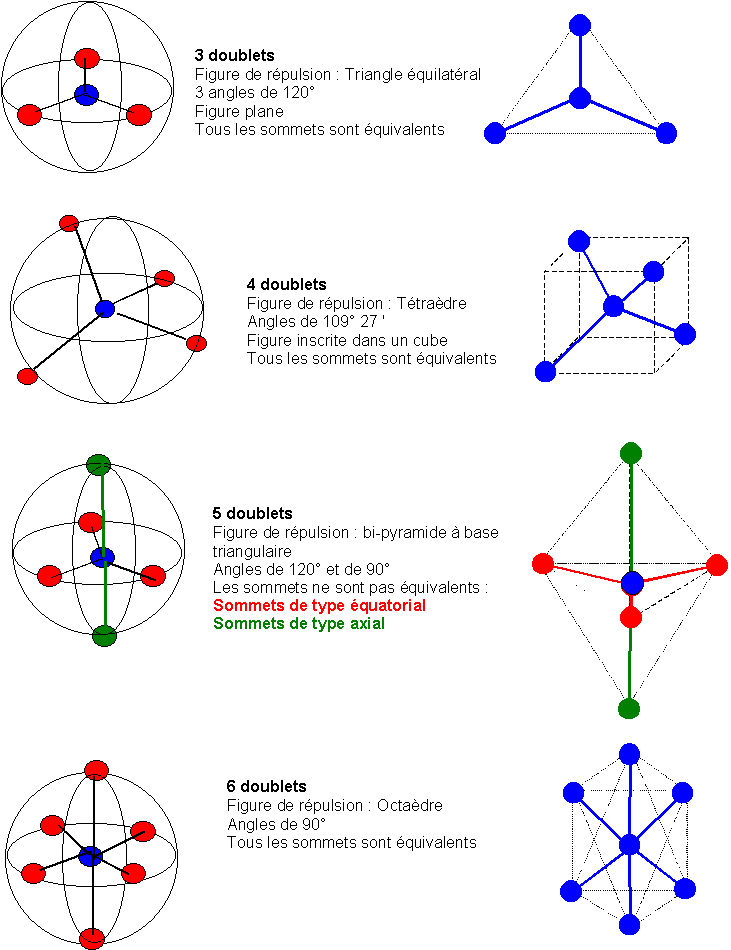
L'hypothèse de Gillespie est que un atome ayant une symétrie sensiblement sphérique, les doublets présents sur la couche de valence vont devoir se répartir à la surface d'une sphère. Cette répartition ne se fait pas au hasard, les doublets électroniques étant chargés négativement se repoussent et vont se placer de manière à être le plus éloigné possible les uns des autres. On obtient ainsi une figure de répulsion différente selon le nombre des doublets présents.
Il existe d'autres figures de répulsions pour un nombre plus élevé de doublets mais nous ne les étudierons pas ici.
La méthode V.S.E.P.R consiste donc à déterminer la position relative des doublets entourant l'atome central, ces doublets pourront être de deux sortes :
Ces deux types de doublets participeront à la figure de répulsion et détermineront la géométrie moléculaire. L'orientation relative des doublets de liaison fixera les directions de ces liaisons et donc la géométrie de la molécule.
Détermination de la géométrie moléculaire :
Le premier travail consiste à trouver le schéma de Lewis moléculaire afin de connaître le nombre de doublets entourant l'atome central et par la même la figure de répulsion correspondante et donc la géométrie moléculaire. Nous avons décrit en détail la manière de procéder pour obtenir ce schéma de Lewis moléculaire. Nous allons distinguer plusieurs types moléculaires conduisant à des géométries différentes, selon le nombre total de doublets entourant l'atome central.
Un type moléculaire sera décrit par le symbolisme suivant : AXnEm
A désigne l'atome central, n est le nombre d'atomes directement liés a l'atome central, m est le nombre de doublets libres de l'atome central.
Reprenons les exemples du chapitre précédent pour illustrer cette codification :
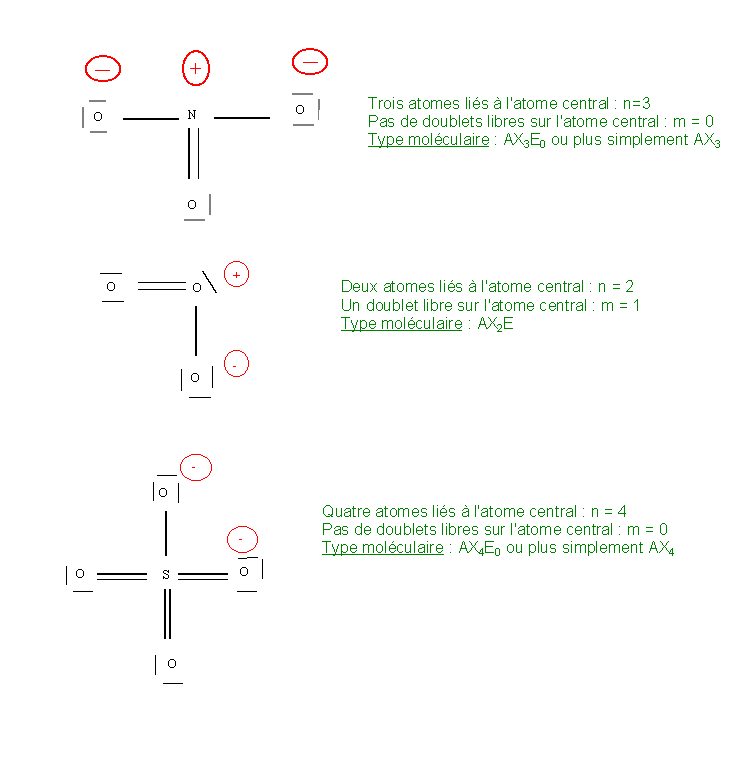
Faisons tout de suite deux remarques importantes :
Une fois le type moléculaire déterminé il suffit de chercher la figure de répulsion associée pour déterminer la géométrie moléculaire. N'oublions pas que c'est le nombre total de doublets qui détermine celle-ci (mis à part la remarque précédante concernant les liaisons multiples). C'est donc la somme p = n + m qui va fixer la géométrie. Selon la valeur de p on aura les figures de répulsions suivantes :
p = 2 : Droite
p = 3 : Triangle équilatéral
p = 4 : Tétraèdre
p = 5 : Bi-pyramide à base triangulaire
p = 6 : Octaèdre
La connaissance de la figure de répulsion n'est pas tout à fait suffisante pour connaître la géométrie de la molécule, en effet les doublets libres participent à la figure de répulsion mais pas directement dans la forme de la molécule qui va être déterminée par l'orientation relative des liaisons. Une molécule de type AX3 avec trois atomes latéraux n'aura évidemment pas la même forme qu'une molécule de type AX2E avec deux atomes latéraux, bien que la figure de répulsion associée soit la même dans les deux cas. Nous allons à présent étudier chaque cas de figure.
1) Trois doublets p = 3
Figure de répulsion : Triangle équilatéral
Types moléculaires : AX3 - AX2E - AXE2
Nous allons représenter ces trois types moléculaires. Les trois sommets étant équivalent la position des doublets libres n'a aucune importance. Les doublets libres seront représentés soit par la lettre E, soit sous formes de lobes.
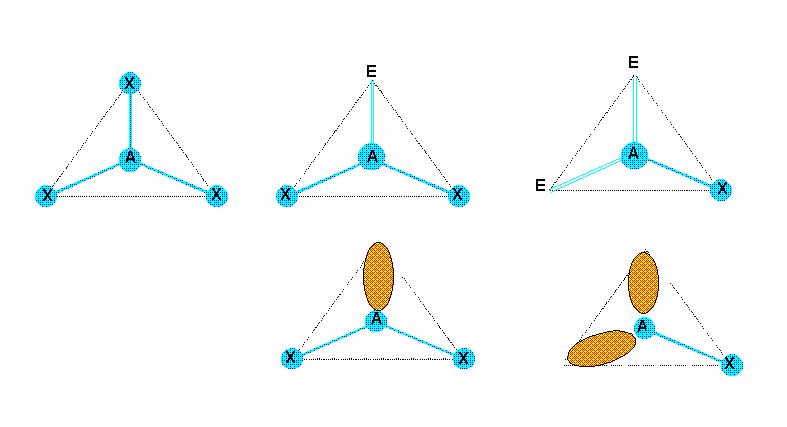
L'examen des figures obtenues montre que :
La molécule AX3 est bien une molécule triangulaire plane.
La molécule AX2E aura la forme d'un V avec un angle de 120°.
La molécule AXE2 sera linéaire.
2) Quatre doublets p = 4
Figure de répulsion : Tétraèdre
Types moléculaires : AX4, AX3E, AX2E2, AXE3
Ici aussi les quatre sommets sont équivalents et la position des doublets libres n'a pas d'importance.
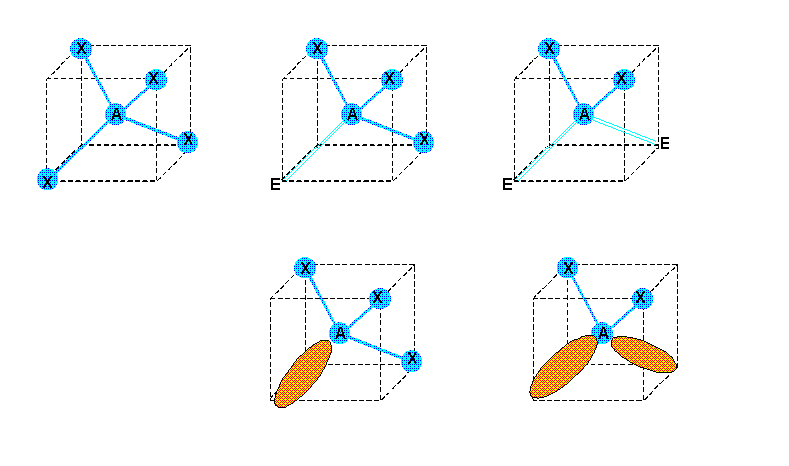
L'examen des figures obtenues montre que :
La molécule de type AX4 est bien tétraédrique
La molécule de type AX3E est en réalité un tétraèdre amputé d'un de ses sommets, la molécule sera donc en fait pyramidale.
La molécule de type AX2E2 aura la forme d'un V avec un angle de 109,5°.
La molécule de type AXE3 (non représentée) sera bien évidemment linéaire.
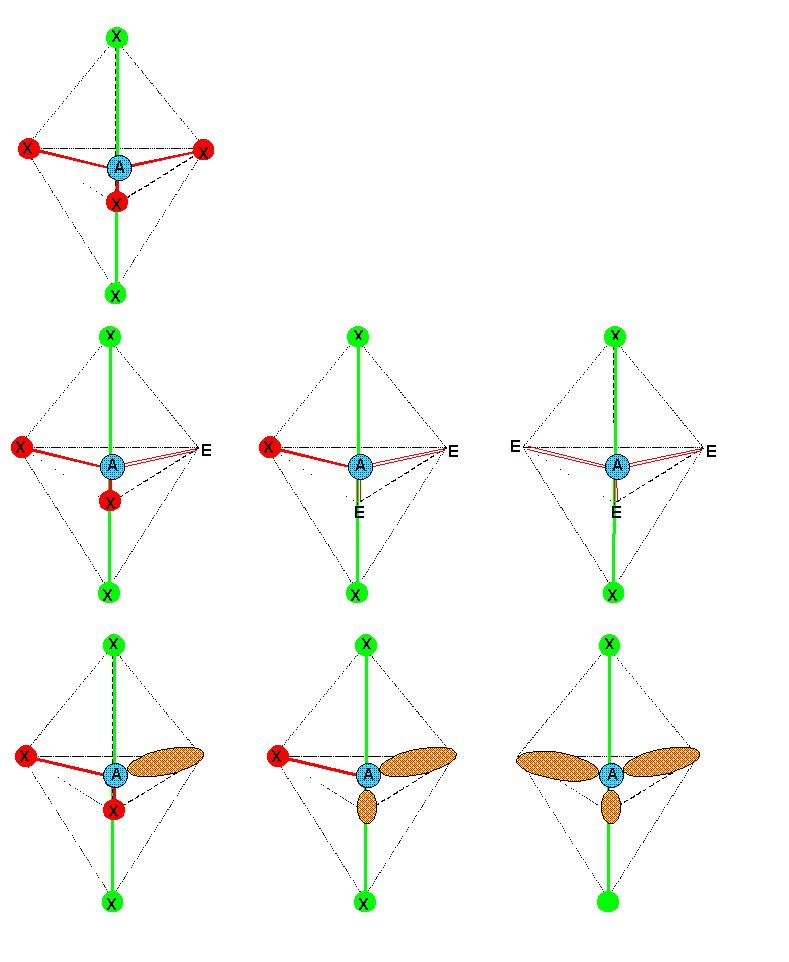
4) 5 doublets p=5
Figure de répulsion : Bi-pyramide à base triangulaire
Types moléculaires : AX5, AX4E, AX3E2,AX2E3,AXE4
Dans ce cas de figure les sommets ne sont plus équivalents et la place relative des doublets libres aura son importance. Pour savoir comment placer ces doublets libres nous admettrons qu'un doublet engagé dans une liaison est "moins encombrant" qu'un doublet libre. Autrement dit un doublet libre occupe un volume plus important qu'un doublet de liaison. Nous reviendrons plus tard sur cette notion d'encombrement. Les doublets libres vont donc se placer sur les sommets qui leur offrent la place la plus grande. Ces sommets sont les sommets équatoriaux, en effet sur de tels sommets offrent des angles de 120° alors que les sommets axiaux n'offrent que des angles de 90°.
La règle que nous retiendrons est donc que pour de telles structures les doublets libres se placeront prioritairement en positions équatoriales.
5) 6 doublets p = 6
Figure de répulsion : Octaèdre
Types moléculaires : AX6, AX5E,AX4E2,AX3E3,AX2E4,AXE5
Les sommets sont tous équivalents. La position du premier doublet libre est donc indifférente. En revanche le deuxième doublet libre se placera obligatoirement à l'opposé du premier. De même, le quatrième doublet libre se placera à l'opposé du troisième. On obtient les figures suivantes :
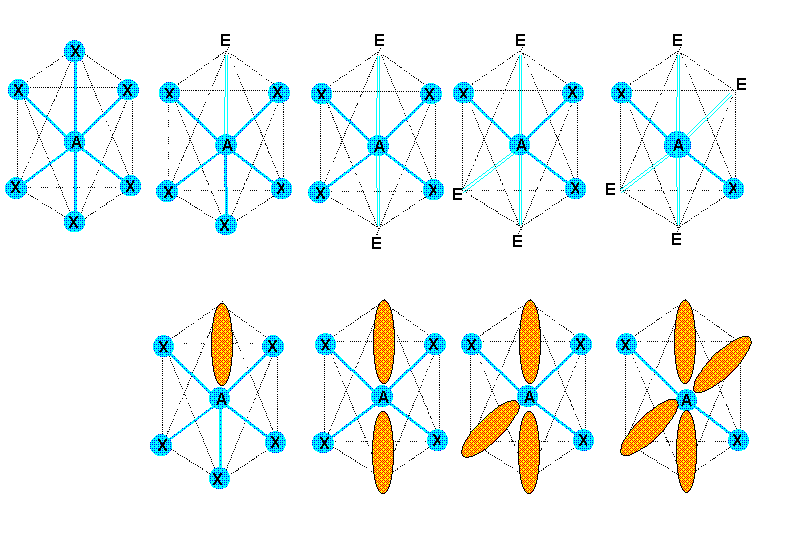
Les figures obtenues montre que :
Le tableau suivant résume tous les cas rencontrés :
Tableau récapitulatif
| p | n | m | type | Figure de répulsion | Géométrie | Angles | Exemples |
| 2 | 2 | 0 | AX2 | Droite | Linéaire | 180 | BeCl2, CO2,HCN |
| 2 | 1 | 1 | AXE | Droite | Linéaire | 180 | |
| 3 | 3 | 0 | AX3 | Triangle équilatéral | Triangle équilatéral | 120 | BF3, AlCl3 |
| 3 | 2 | 1 | AX2E | Triangle équilatéral | Coudée Forme de V | 120 | SO2,SnCl2, O3 |
| 3 | 1 | 2 | AXE2 | Triangle équilatéral | Linéaire | 180 | |
| 4 | 4 | 0 | AX4 | Tétraèdre | Tétraèdre | 109,5 | CH4, NH4+, SO42- |
| 4 | 3 | 1 | AX3E | Tétraèdre | Pyramide déformée | 109,5 | NH3, H3O+ |
| 4 | 2 | 2 | AX2E2 | Tétraèdre | Coudée Forme de V | 109,5 | H2O, H2S |
| 4 | 1 | 3 | AXE3 | Tétraèdre | Linéaire | 180 | |
| 5 | 5 | 0 | AX5 | Bi-pyramide triangle | Bi-pyramide triangle | 120 et 90 | PCl5 |
| 5 | 4 | 1 | AX4E | Bi-pyramide triangle | Pyramide déformée | 120 et 90 | SF4, TeCl4 |
| 5 | 3 | 2 | AX3E2 | Bi-pyramide triangle | Forme de T | 90 | ICl3, ClF3 |
| 5 | 2 | 3 | AX2E3 | Bi-pyramide triangle | Linéaire (3 atomes) | 180 | I3-, XeF2, ICl2- |
| 5 | 1 | 4 | AXE4 | Bi-pyramide triangle | Linéaire (2 atomes) | 180 | |
| 6 | 6 | 0 | AX6 | Octaèdre | Octaèdre | 90 | SF6 |
| 6 | 5 | 1 | AX5E | Octaèdre | Pyramide carrée | 90 | BrF5, IF5 |
| 6 | 4 | 2 | AX4E2 | Octaèdre | Carrée (plane) | 90 | XeF4, BrF4- |
| 6 | 3 | 3 | AX3E3 | Octaèdre | Forme de T | 90 | |
| 6 | 2 | 4 | AX2E4 | Octaèdre | Linéaire | 180 |
Améliorations du modèle :
Le modèle précédent est très simple et permet de prévoir avec un très bonne approximation les géométries moléculaires. Si on compare les prévisions du modèle avec les géométries déterminées expérimentalement on note toutefois des différences sensibles. La plupart de ces différences sont néanmoins facilement explicables par de petites améliorations du modèle.
La méthode V.S.E.P.R comme nous l'avons dit est basée sur la répulsion qu'exercent entre eux les doublets entourant l'atome central. D'après la loi de Coulomb (F = K * q q' / d2)cette répulsion dépend de deux facteurs, les charges électriques et la distance qui les séparent. La répulsion sera donc d'autant plus élevée que les charges seront importantes ou la distance les séparant faible.
Nous n'avons jusqu'ici pas tenu compte de ces facteurs en supposant que tous les doublets étaient identiques et situés à la même distance de l'atome central (symétrie globale sphérique).
Nous allons maintenant tenir compte de ces facteurs pour affiner le modèle.
Influence des liaisons multiples :
Les liaisons doubles correspondent à deux doublets. Ces liaisons correspondent donc à des charges plus élevées que les liaisons simples, cela va correspondre à des répulsions plus élevées qui vont modifier les angles de la figure de répulsion. La présence d'une liaison double va donc modifier légèrement la géométrie moléculaire.
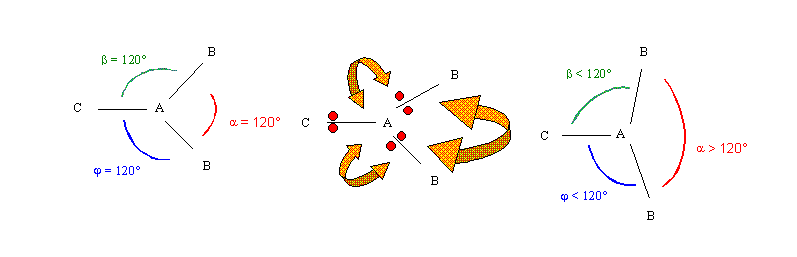
Prenons un exemple pour illustrer cet effet. Soit la molécule hypothétique AXX'X", de schémas de Lewis moléculaire AX3 présentant une liaison double et deux liaisons simples. Nous ne nous préoccuperons pas pour l'instant de la nature des atomes X, ,X' et X" mais seulement de l'effet de la double liaison. Théoriquement les angles devraient être égaux à 120°. Mais en réalité la présence de la double liaison va les modifier. La double liaison correspondant à deux doublets va créer une répulsion plus forte qui va faire augmenter les deux angles pour lesquels elle est concernée et faire diminuer le troisième angle. Une figure le montrera mieux qu'un discours, les points rouges symbolisent les électrons, les flèches oranges les répulsions (la flèche est d'autant plus large que la répulsion est importante).
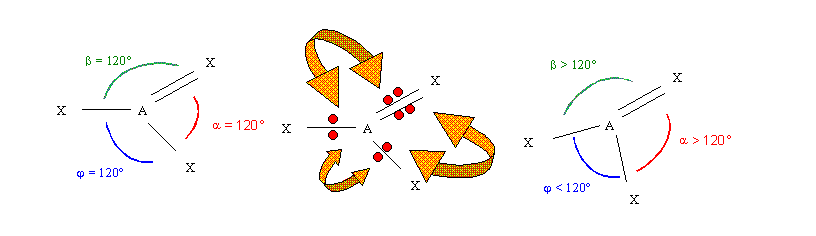
Nous retrouverons cet effet chaque fois qu'une double liaison sera présente. Pour des raisons qui ne seront expliquées que plus tard, les triples liaisons impliquent une géométrie linéaire et n'influencent donc pas les angles.
Influence des doublets libres :
La nature différente des doublets de liaisons (X) et des doublets libres (E) va, elle aussi modifier légèrement la géométrie moléculaire. Nous avons d'ailleurs implicitement tenu compte de cet effet quand nous avons étudié les structures à 5 doublets (AX5, AX4E, AX3E2, AX4E) en affirmant que les doublets libres étaient plus volumineux que les doublets de liaison. Nous allons maintenant expliquer ce phénomène. C'est ici la distance entre les doublets qui va entraîner une modification de la figure de répulsion. En effet les doublets engagés dans une liaison sont soumis à l'influence de l'atome latéral. Ils sont attirés par le noyau chargé positivement de cet atome et vont donc se trouver plus éloignés de l'atome central que les doublets libres. L'éloignement de deux doublets de liaisons sera plus élevé que celui entre deux doublets libres ou celui entre un doublet libre et un doublet de liaison. Comme la répulsion est d'autant plus élevée que l'éloignement est faible l'effet répulsif des doublets libres sera toujours plus élevé que celui des doublets de liaisons. Comme précédemment cela va se traduire par une modification des angles. Reprenons un exemple du même type que le précédant. Soit une structure du type AX2E, la molécule devrait avoir la forme d'un V avec un angle de 120°.L'effet du doublet libre va modifier cet angle en le faisant diminuer.
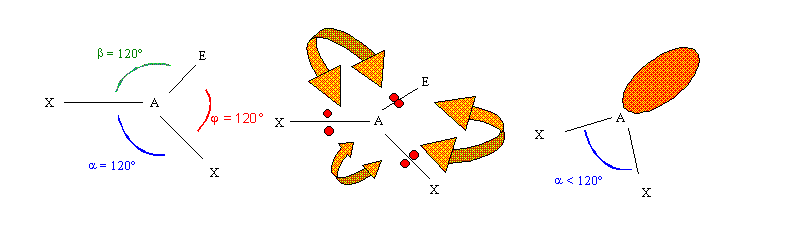
Cet effet sera d'autant plus important que le nombre de doublets libres augmentera. Par exemple les molécules CH4, NH3 et H2O présentent toutes une figure de répulsion tétraédrique respectivement AX4, AX3E et AX2E2 on devrait donc observer des angles de 109,5° pour toutes les trois. En réalité seul CH4 présente cette symétrie tétraédrique parfaite. Dans NH3 les angles HNH sont de 107,6° (effet de 1 doublet libre), dans H2O la valeur de l'angle HOH diminue encore pour passer à 104,5° (effet de 2 doublets libres).
Influence de la nature des atomes latéraux :
La nature des atomes latéraux va, elle aussi, influer sur la géométrie moléculaire. C'est par leur électronégativité qu'ils vont modifier la distance entre les doublets de liaisons et l'atome central et comme précédemment modifier les angles de la figure de répulsion. Supposons une molécule AB2C, de type AX3. Supposons que l'électronégativité de A soit supérieure à celle de B et que celle de C soit inférieure à celle de A. Plus un atome est électronégatif et plus il attire les électrons à lui les doublets de liaisons AC seront donc attirés vers C, ceux des liaisons AB seront attirés par A. Comme le montre la figure les doublets AB seront proches et exerceront une forte répulsion entre eux. A l'opposé les répulsion entre doublets AB et AC plus éloignés seront plus faibles. Au final, les angles BAC auront diminué alors qu'au contraire l'angle BAB augmentera.
Influence de l'atome central :
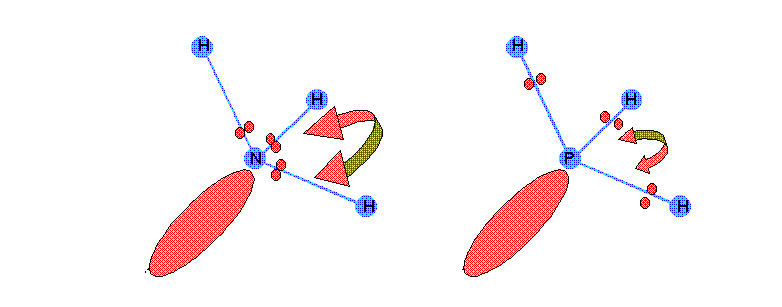
Pour des molécules ne différant que par la nature de l'atome central on observera aussi des variations d'angles. Ces variations s'expliquent de la même manière que précédemment par l'effet des différences d'électronégativité entre l'atome central et les atomes latéraux. Par exemple NH3 et PH3 sont deux molécules de type AX3E ne différant que par la nature de l'atome central. Ces deux molécules présenteront néanmoins des angles XAX de valeurs différentes.
Les électronégativités d'Alred et Rochow des éléments concernés sont les suivantes :
XH = 2,2 - XN = 3 - XP = 2
En raison des électronégativités différentes les doublets seront près de N dans NH3 et à l'inverse près de H dans PH3 . Les répulsions seront donc plus fortes dans NH3 que dans PH3 et l'angle HNH sera plus grand que l'angle HPH.
Conclusion :
Bien entendu toutes les influences particulières étudiées se conjuguent dans les molécules réelles.
Il sera donc très difficile et souvent impossible de prévoir les valeurs exactes des divers angles. Néanmoins la méthode V.S.E.P.R de par sa simplicité est une méthode irremplaçable qui permet d'obtenir une bonne approximation de ces angles.
LA LONGUEUR DES LIAISONS :
On peut attribuer une certaine longueur aux liaisons chimiques unissant les atomes entre eux.
Ces longueurs de liaisons sont même accessibles expérimentalement. Leur ordre de grandeur est l'Angström (Å) soit 10-10 m. La longueur d'une liaison entre deux atomes n'est pas une constante absolue. On peut néanmoins assigner une valeur moyenne à chaque longueur de liaison. La valeur exacte dépendra en partie de l'environnement, c'est à dire des autres liaisons présentes dans la molécule étudiée.
Utilisation des rayons de covalence des atomes :
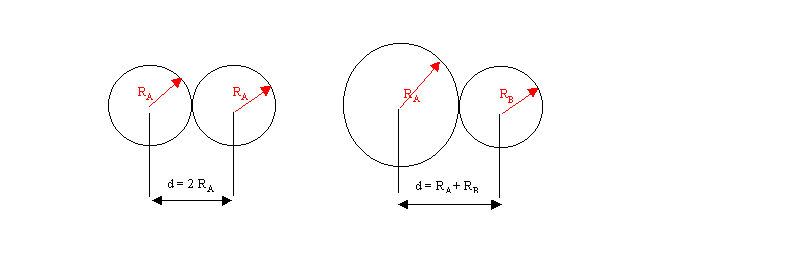
Si on assimile les atomes à des sphères on peut assigner un certain rayon à chaque atome. La grandeur la plus utilisée parce que facile à définir est le rayon de covalence.
Nous l'avons définit lors de l'étude des atomes comme étant la moitié de la longueur de la simple liaison unissant deux atomes identiques. Connaissant les rayons de covalence de deux atomes (ces valeurs sont souvent indiquées dans les classifications périodiques) on pourra estimer la longueur d'une liaison A-B par la somme des rayons de covalence des deux atomes concernés. Le résultat obtenu n'est pas toujours excellent car la longueur d'une liaison dépende de l'environnement et n'est donc pas rigoureusement constante. Cette méthode simple permet néanmoins d'obtenir une assez bonne approximation des longueurs de liaisons.
Il faut bien entendu disposer au préalable des rayons de covalence expérimentaux de chaque atome pour pouvoir l'utiliser.
Un facteur modifiant sensiblement la longueur des liaisons est la différence d'électronégativité des atomes concernés. Une différence d'électronégativité importante (liaison polarisée) entraîne un raccourcissement de la liaison. Une formule empirique a été proposée pour tenir compte de cet effet et corriger la longueur de liaison calculée :
d = RA + RB - 9 DX
Dans cette formule DX représente la différence d'électronégativité (en valeur absolue) entre les atomes A et B. L'échelle d'électronégativité utilisée est l'échelle de Pauling. L'unité de longueur utilisée est ici le picomètre (1pm = 10-12 m)
Comme précédemment l'utilisation de cette formule corrigée nécessite la connaissance préalable des rayons de covalence ainsi que celle des électronégativités de Pauling des atomes concernés.
Un deuxième facteur important est la multiplicité de la liaison. Une liaison sera d'autant plus courte que sa multiplicité sera élevée.
Exemples :
CC simple d = 1,54 Å ; CC double d = 1,47 Å ; CC triple d = 1,43 Å
CN simple d = 1,47 Å ; CN double d =1,30 Å ; CN triple d = 1,16 Å
Méthode ne nécessitant pas la connaissance préalable des rayons de covalence :
Il serait intéressant de pouvoir calculer à priori la longueur d'une liaison sans avoir à connaître au préalable les valeurs des rayons de covalence des atomes données par les tables.
Cela est possible par l'utilisation de la méthode suivante que j'ai personnellement mise au point (voir article paru dans le Bulletin de l'Union des Physiciens Vol 91 - juin 97).
Le calcul à priori des rayons de covalence (en Å) est possible avec une assez bonne approximation en utilisant la formule suivante :
R (Å )= 0,215 n*2/Z* + 0,148 n* + 0,225
Z* est la charge ressentie par un électron de la couche de valence de l'atome étudié calculée par les règles de Slater
n* est la valeur corrigée du nombre quantique principal de la couche de valence de l'atome étudié.
Avec n* = n pour n = 2 ou n = 3 ; n* = 3,6 pour n=4 et n* = 4 pour n = 5
On pourra donc utiliser cette formule pour déterminer à priori les rayons de covalence des atomes concernés et calculer ensuite la longueur approximative Lcalc de la liaison étudiée.
On pourra améliorer la précision en utilisant la formule suivante :
L (Å ) = 1,11 Lcalc - 0,203
On peut éviter le calcul intermédiaire des rayons de covalence par l'utilisation de la formule suivante :
L (Å ) = 0,239 S(n*2 / Z*) + 0,164 S(n*) + 0,297
L'accord obtenu est assez bon, l'écart moyen entre les valeurs calculées par cette formule et les valeurs expérimentale étant de 1,5 %.
L'atome d'hydrogène est un cas particulier on donnera la valeur de 0,346 Å à son rayon de covalence.
Le calcul des longueurs de liaisons multiples est possible car il existe un rapport pratiquement constant entre la longueur des liaisons simples et multiples.
La longueur de la liaison double est de 86 % de celle de la simple.
La longueur de la liaison triple est de 78% de celle de la simple.
Un écart moyen de 2% est obtenu pour les liaisons multiples.